





- Preparation and Properties of Polyurethane Using Methoxylated Vegetable Oil as Chain Extender
Hyung Woo Koo, Young Jun Kim†
 , and Eun-Soo Park*,†
, and Eun-Soo Park*,† Department of Food & Biotechnology, Korea University Sejong Campus, 2511, Sejong-ro, Jochiwon-eup, Sejong-si 30019, Korea
*Intechnology Co., Ltd., 861-46, Poseungjangan-ro, Jang-an-ri, Jangan-myeon, Hwaseong-si, Gyeonggi-do 18586, Korea- 사슬연장제로 메톡실화 식물성 오일을 이용한 폴리우레탄의 제조 및 물성
구형우 · 김영준†
· 박은수*,† 고려대학교 세종캠퍼스, *(주)인테크놀로지
Reproduction, stored in a retrieval system, or transmitted in any form of any part of this publication is permitted only by written permission from the Polymer Society of Korea.
A series of polyurethane (PU) elastomers were synthesized using methoxylated vegetable oils as chain extenders to investigate the effects of their content on the mechanical properties and biodegradability of the resulting PUs. The tensile strength of the chain-extended PU exhibited a maximum at 15 wt% of the methoxylated vegetable oil content, followed by a significant decrease. Conversely, the hardness gradually decreased with increasing content of the methoxylated vegetable oil. Biodegradation tests using Pseudomonas aeruginosa E7 revealed that the chain-extended PU with 15 wt% of methoxylated sunflower oil (MSR), methoxylated corn oil (MCN), methoxylated castor oil (MCR), and methoxylated canola oil (MCA) exhibited biodegradation rates of 16.1, 15.2, 14.2, and 13.6%, respectively, without any initial lag phase.
메톡실화된 식물성 기름을 사슬연장제로 사용하여 일련의 폴리우레탄(PU) 엘라스토머를 합성하고, 이들의 함량이 PU의 기계적 성질 및 생분해성에 미치는 영향을 조사하였다. 사슬연장된 PU의 인장강도는 메톡실화된 식물성 기름의 함량이 15 wt%일 때 최대치를 나타낸 후, 현저히 감소하였다. 반대로, 경도는 메톡실화된 식물성 기름의 함량이 증가함에 따라 점진적으로 감소하였다. 녹농균 E7을 이용한 생분해 시험 결과, methoxylated sunflower oil(MSR), methoxylated corn oil(MCN), methoxylated castor oil(MCR) 및 methoxylated canola oil(MCA)를 15 wt% 함유하는 사슬연장된 PU는 초기 지연 없이 각각 16.1, 15.2, 14.2 및 13.6%의 생분해율을 나타내었다.
Lack of degradability and the various pollution problems have led to increasing concern about synthetic polymers. In this study, a series of polyurethane elastomers were synthesized using methoxylated vegetable oils as chain extenders to investigate the effects of their content on the mechanical properties and biodegradability of the resulting polyurethanes.

Keywords: chain extender, biodegradation, elastomer, epoxidation, polyurethane.
The
authors declare that there is no conflict of interest.
The environmental impact of non-degradable synthetic polymers, coupled with dwindling fossil fuel resources, has spurred interest in sustainable alternatives.1 As the demand for eco-friendly products grows, the development of biodegradable and recyclable polymers has become increasingly important.2
Naturally occurring vegetable oils offer a sustainable and cost-effective renewable resource, providing a versatile starting point for developing innovative products with diverse functional and structural properties.1 Especially epoxidation of them is a common technique for producing epoxides, highly reactive compounds that readily undergo ring-opening reactions under acidic conditions. This process is frequently employed in the synthesis of diols and polyols, which serve as valuable precursors for a wide range of hydroxyl-functional compounds and polymers.3
Polyurethane (PU), a versatile man-made polymer derived from the reaction between polyisocyanates and polyols, is used in a wide variety of products in the medical, automotive, food, and industrial fields.4-6 However, its high resistance to degradation leads to environmental accumulation. This persistent waste poses risks to human health and ecosystems. Traditional disposal methods like landfill and incineration harm the environment. Biodegradation is emerging as a sustainable alternative for PU waste treatment.4
In this study, a series of biodegradable PU were obtained by chain extension of prepolymer with the addition of methoxylated vegetable oils. The vegetable oils used in this study were castor oil (CR), sunflower oil (SR), corn oil (CN), and canola oil (CA) (Scheme 1). The influence of the chain extender concentration on the mechanical properties and biodegradability of the prepared PU was evaluated. The epoxidation of four vegetable oils was carried out using peroxyacid generated in-situ by reacting concentrated hydrogen peroxide (H2O2) with formic acid. Methoxylated vegetable oil polyols were then synthesized by ring opening of epoxidized vegetable oils in the presence of fluoboric acid (Scheme 2).7 Biodegradability tests were performed in liquid medium using Pseudomonas aeruginosa E7 (P. aeruginosa E7), isolated from petroleum-contaminated soil.8
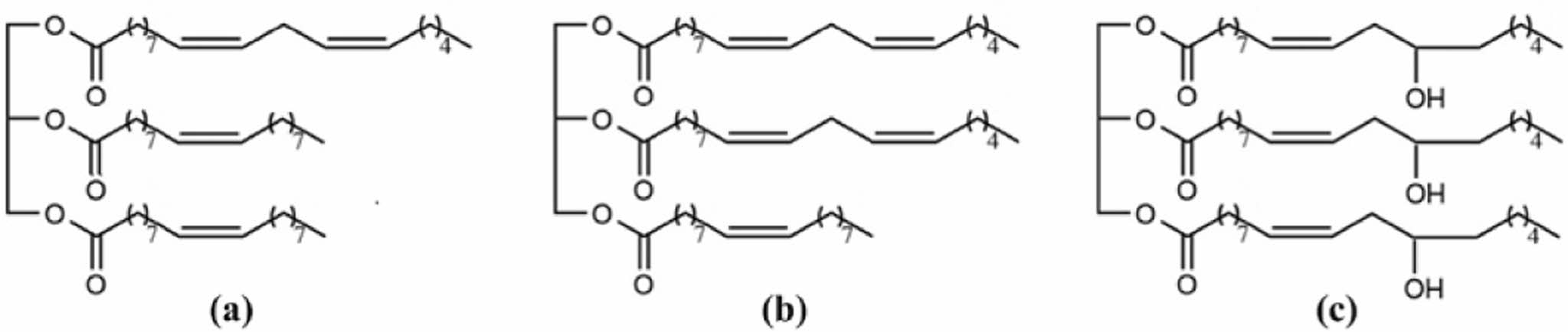
Scheme 1. Idealized chemical structures of the (a) CA; (b) SR; (b) CN; (c) CR.
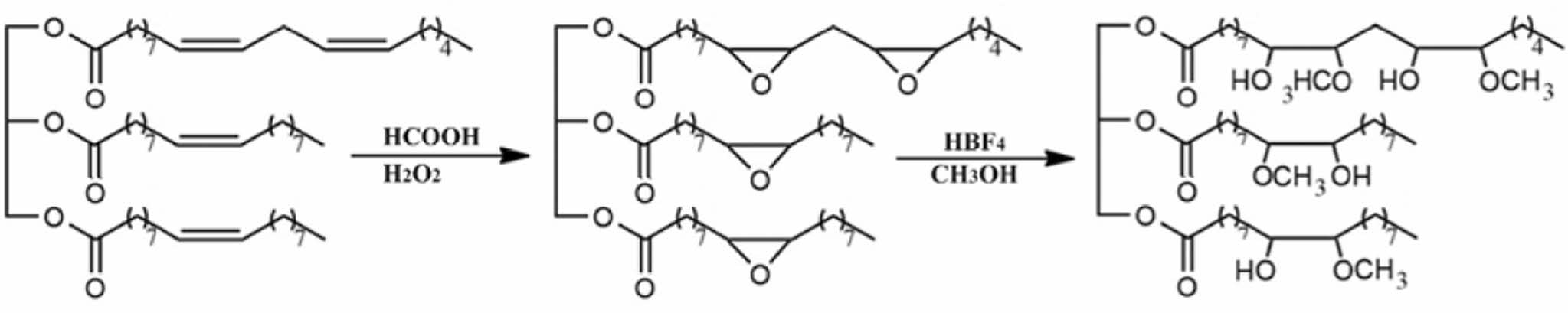
Scheme 2. Idealized schematic diagram of the epoxidation and methoxylation of CA.
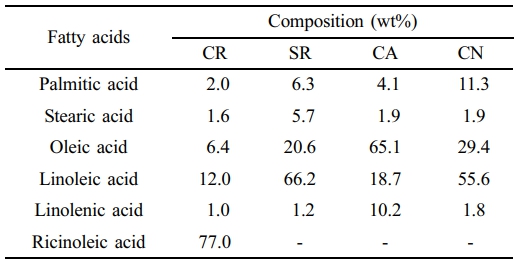
Materials. CN, SR, and CA oils were manufactured by Haepyo, Korea and purchased from grocery stores. CR was extracted from the seeds of the Castor plant. Fluoboric acid, poly(tetramethylene ether) glycol (PTMG, Mw » 2000), 1,4-phenylene diisocyanate (PDI), and tin(II) 2-ethyl-hexanoate (TEH) were supplied by Sigma-Aldrich. Table 1 summarizes the percent fatty acid composition by weight of four vegetable oils.
Instrumentation. Proton nuclear magnetic resonance (1H NMR) spectra recorded at room temperature on a Bruker AC-250 Fourier transform nuclear magnetic resonance (FT-NMR, Germany) spectrometer and analyzed using commercial software.
The surface of the cryogenic-fractured PU sample was measured using a Hitachi S-4300 (Japan) scanning electron microscope (SEM).
Fourier transform infrared (FTIR) spectra of oil samples were measured in the wavenumber range of 4000-400 cm-1 using a PerkinElmer Spectrum 2000 IR spectrometer (USA) and analyzed using commercial software.
The tensile test of the IEC 60811-1-1 type dumbbell specimen was performed at a crosshead speed of 50 mm/min using a DEC-A500TC (Dawha Test Machine, Korea) universal testing machine (UTM).
Thermal decomposition of oil samples was determined by Q50 thermogravimetric (TG) analyzer (TA instrument, USA).
Epoxidation of Vegetable Oil. H2O2 (12 g) was added dropwise to a mixture of vegetable oil (50 g) and formic acid (12 g) over 5 h at 50 ℃. The reaction mixture was stirred for an additional 5 h, then diluted with ethyl acetate and water. The aqueous layer was separated, and the organic layer was concentrated under reduced pressure.
Synthesis of Methoxylated Vegetable Oil. Epoxidized vegetable oil (10 g) was added dropwise over 10 min to a mixture of fluoboric acid (0.4 g), methyl alcohol (10 mL), water (1 g), and isopropyl alcohol (30 mL) maintained at 40 ℃. The mixture was stirred at 50 ℃ for 1 h, at which stage ammonium hydroxide (0.6 mL) was added to stop the reaction. Then concentrate the reaction mixture using a rotary evaporator under reduced pressure.
Preparation of PU. PDI (8 g, 0.05 mol), PTMG (45 g, 0.0225 mol), TEH (0.05 mL), and toluene (50 g) were added to a reactor equipped with a reflux condenser, nitrogen inlet, and mechanical stirrer. The reaction mixture was stirred continuously at 80 rpm for 2 h at 60 ℃, yielding a light yellow, low-viscosity liquid (Scheme 3).
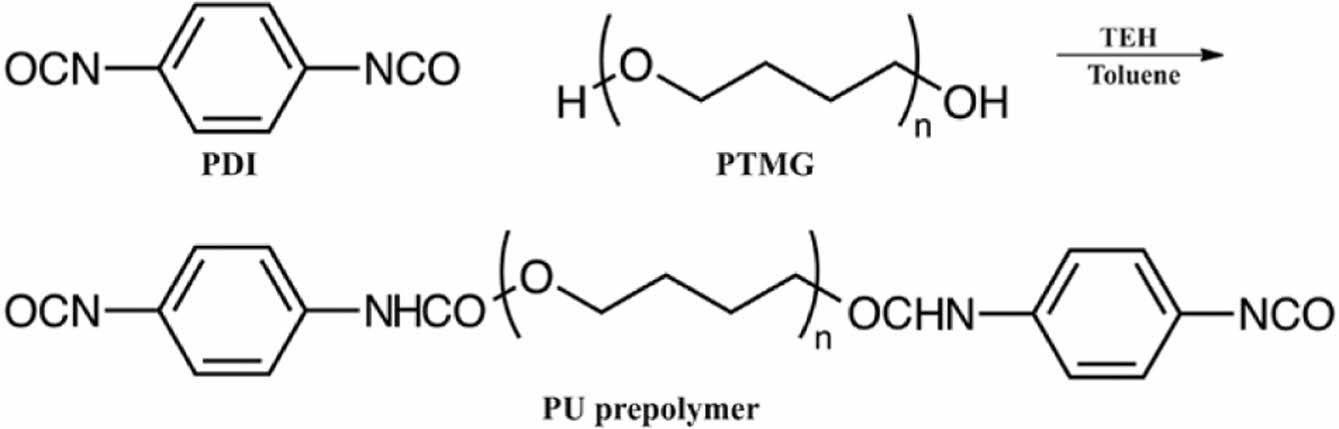
Scheme 3. Preparation of the PU prepolymer.
To prepare the PU formulations, 11.12 (10 wt%), 17.65 (15 wt%), and 25.00 (20 wt%) parts by weight of each methoxylated vegetable oil were premixed with 100 parts by weight of prepolymer, followed by the addition of 0.32 parts of TEH. The resulting mixtures were stirred for 10 min, poured into glass Petri dishes, and cured at 60 ℃ for 2 h.
Modified Sturm Test. Biodegradation of the PU samples was assessed using the Modified Sturm test (ASTM D 5209-91) at 37 ℃ and 100 rpm. CO2, a byproduct of microbial degradation of the samples by P. aeruginosa E7, was absorbed by 0.025 N barium hydroxide and quantified through titration with 0.05 N hydrochloric acid.8
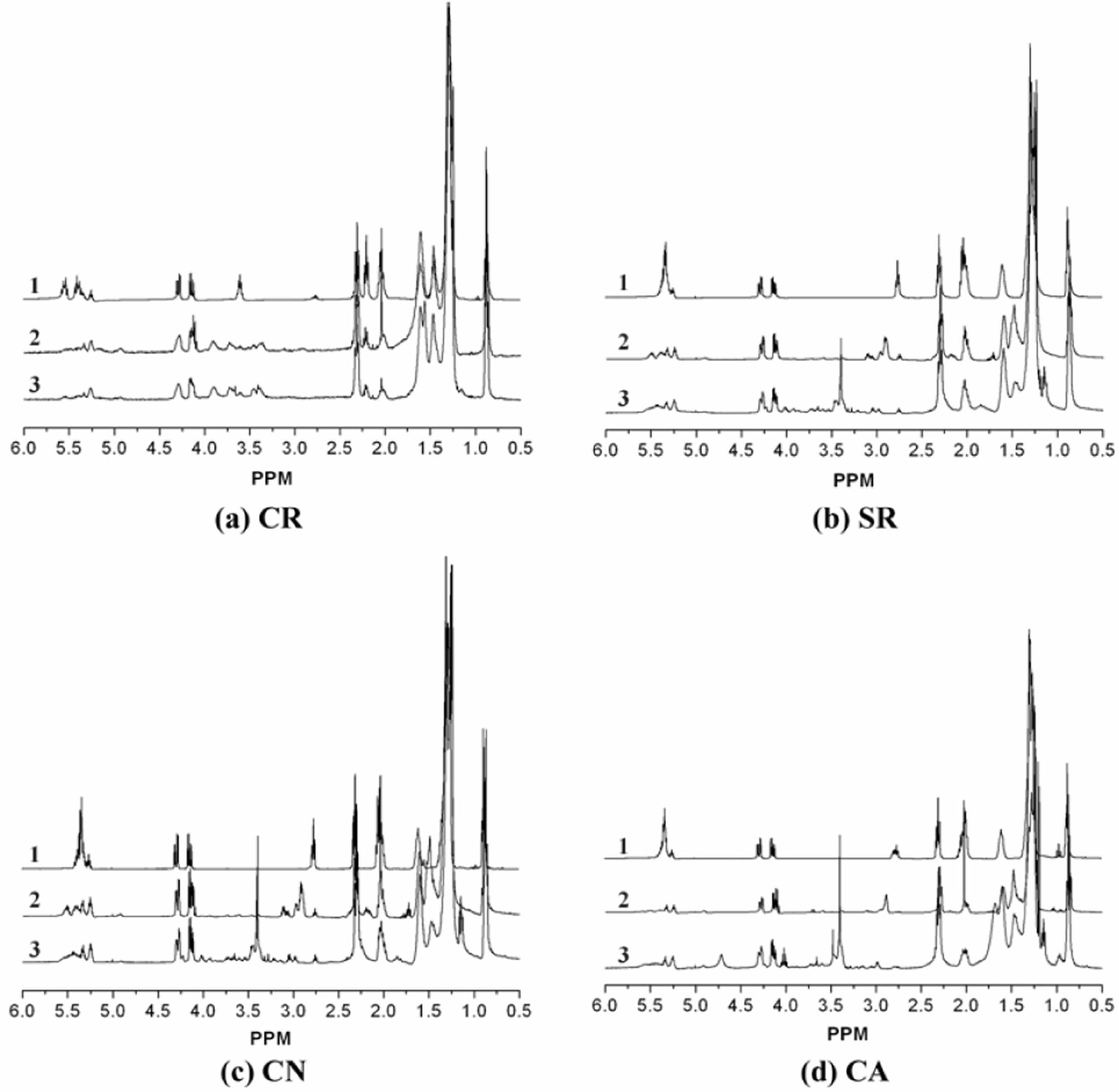
Synthesis of Epoxidized- and Methoxylated Oils. Figure 1 displays the 1H NMR spectra of the pure oil (Figure 1-1), epoxidized oil (Figure 1-2), and methoxylated oil (Figure 1-3) at ambient temperature. The 1H NMR spectrum shows peaks at 0.87 and 1.25 ppm, corresponding to terminal methyl and methylene groups of fatty acid chains, respectively. Allylic protons of fatty acid chains appear at 2.0 ppm, while protons adjacent to ester linkages are observed at 2.3 ppm.1 Additionally, peaks at 4.1-4.3 and 5.3-5.6 ppm are assigned to methylene groups in the glyceride unit and unsaturated carbons, respectively.
Following the procedure outlined in Scheme 2, four vegetable oils were epoxidized and subsequently methoxylated. Upon epoxidation, the distinctive 1H NMR peaks (5.3-5.6 ppm) corresponding to olefinic protons in four oils were significantly attenuated (Figure 1-1), while new peaks emerged at 2.8-3.2 ppm (Figure 1-2), characteristic of newly formed epoxy groups.
1H NMR spectral analysis of the epoxidized and methoxylated oils revealed a significant structural change. The disappearance of the epoxy group peak centered around 2.8-3.2 ppm (Figure 1-2), coupled with the appearance of a new peak at 3.4 ppm (Figure 1-3) corresponding to the methoxy group's methyl ester protons, confirms the conversion of the epoxy ring to a methoxy group.1 The epoxy group content and degree of unsaturation were quantified using 1H NMR spectroscopy.
Based on 1H NMR analysis, the residual unsaturation per molecule was determined to be 0.1 for the methoxylated CR (MCR) and 0.2 for the methoxylated CA (MCA). The methoxylated SR (MSR) and methoxylated CN (MCN) exhibited similar amounts of residual unsaturation (1.9 and 1.8, respectively). The MSR, MCN, MCA, and MCR under investigation had approximately 2.5, 2.5, 2.2, and 2.7 hydroxyl groups per molecule, respectively.
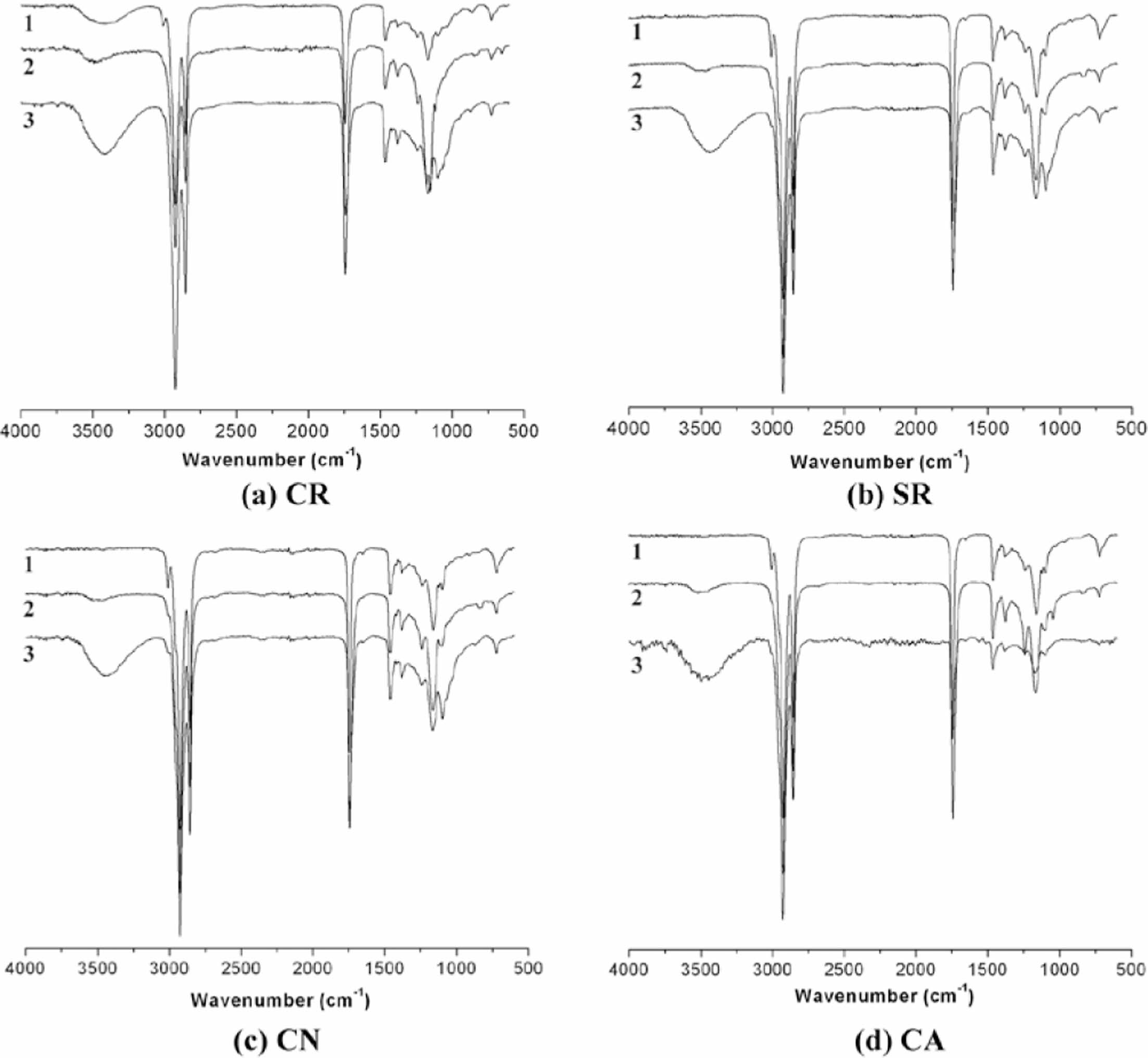
FTIR spectroscopy was employed to further characterize the epoxidized and methoxylated vegetable oils, and the results are presented in Figure 2. The FTIR spectrum of the pure oils (Figure 2-1) exhibits characteristic peaks at 3008, 1650, and 720 cm-1, corresponding to the stretching vibrations of =C-H, -C=C-, and -CH=CH-, respectively.9 Following epoxidation (Figure 2-2) at 50 ℃ for 10 h, the peak at 3008 cm-1, indicative of double bonds, nearly vanished. Additionally, the emergence of a new peak at 831 cm-1, attributed to the epoxy group, confirmed the successful epoxidation of the four vegetable oils.
Compared to the FTIR spectrum of epoxidized oils, the FTIR spectra of the methoxylated oils (Figure 2-3) exhibited the disappearance of the epoxy group peak and the emergence of a hydroxyl group peak at approximately 3470 cm-1, indicative of ring-opening reaction. From Figure 2b-2, 2c-2, and 2d-2, the hydroxyl functional groups were observed in the epoxy products. The presence of the hydroxyl group peak in the spectra of the epoxidized products suggests that partial epoxy ring-opening reactions may have occurred. The presence of water from the aqueous H2O2 solution used during epoxidation could have facilitated acid-catalyzed ring-opening reactions.10
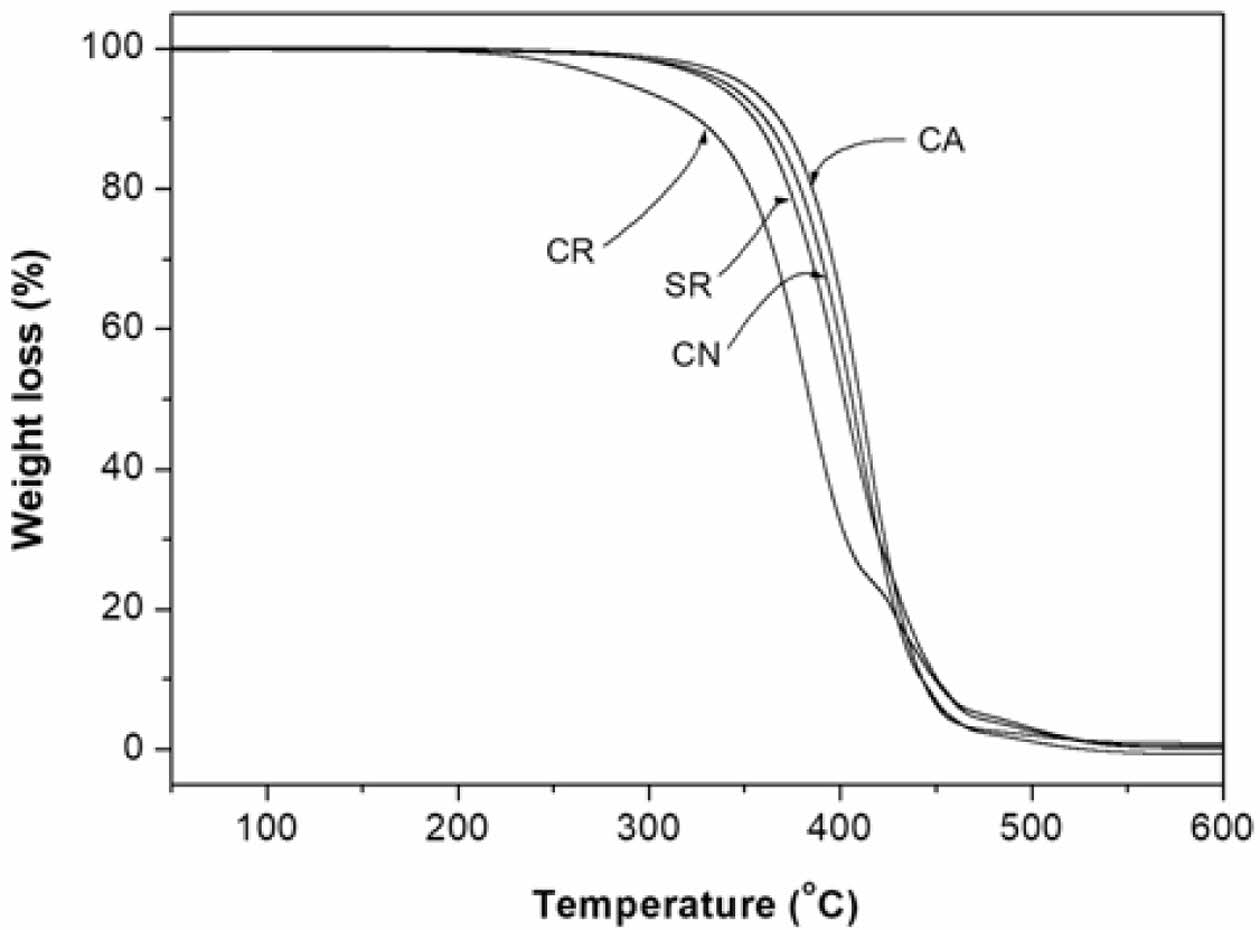
Thermal Degradation of Pure, Epoxidized- and Methoxylated Oils. As shown in Figure 3, the TGA curves of CN, SR, and CA have similar characteristics with two thermal decomposition steps in the range of 320 to 550 ℃, and no residue remains at 800 ℃. Typically, vegetable oil thermal degradation involves three phases: polyunsaturated fatty acid breakdown (230-380 ℃), monounsaturated fatty acid decomposition (380-480 ℃), and saturated fatty acid degradation (480-550 ℃).11 However, the thermal decomposition of CN, SR, and CA occurred in only two steps (approximately 286-460 ℃ and 460-550 ℃) likely due to the superposition of the steps. Conversely, CR exhibits a three-step degradation pattern. The initial decomposition stage commences at 210 ℃, followed by a rapid weight loss phase extending to 460 ℃. The final stage, spanning 460-550 ℃, involves the complete decomposition of the intermediate pyrolysis product.11
Based on the weight loss at 320 ℃ obtained from the TGA curves, it can be established that CA (98.0 %) had a higher thermal stability than that observed for CN (97.2%), SR (96.3%), and CR (90.5%). The thermal instability of vegetable oils is attributed to the presence of double bonds in the fatty acid chains and the β-hydrogen atoms in the glycerol backbone. The double bonds are particularly reactive and readily undergo oxidation reactions with atmospheric oxygen, while the β-hydrogen atoms are easily abstracted, leading to the cleavage of ester linkages and the formation of olefins and acids.12
Thus, the thermal stability of CN, SR, and CA is dependent on the composition of the fatty acids. The low concentrations of linolenic acid besides the high content of oleic acid assure thermal stability to the CA. Highly polyunsaturated CN and SR are prone to rapid deterioration at elevated temperatures.
The low thermal stability of CR may be due to the presence of a secondary hydroxyl group of ricinoleic acid, besides the presence of small amounts of oleic acid. This indicates that hydroxyl moieties in unsaturated ricinoleic acid may impart mild pro-oxidant behavior to CR. The secondary hydroxyl group is thermally unstable and easily undergoes dehydration.13,14
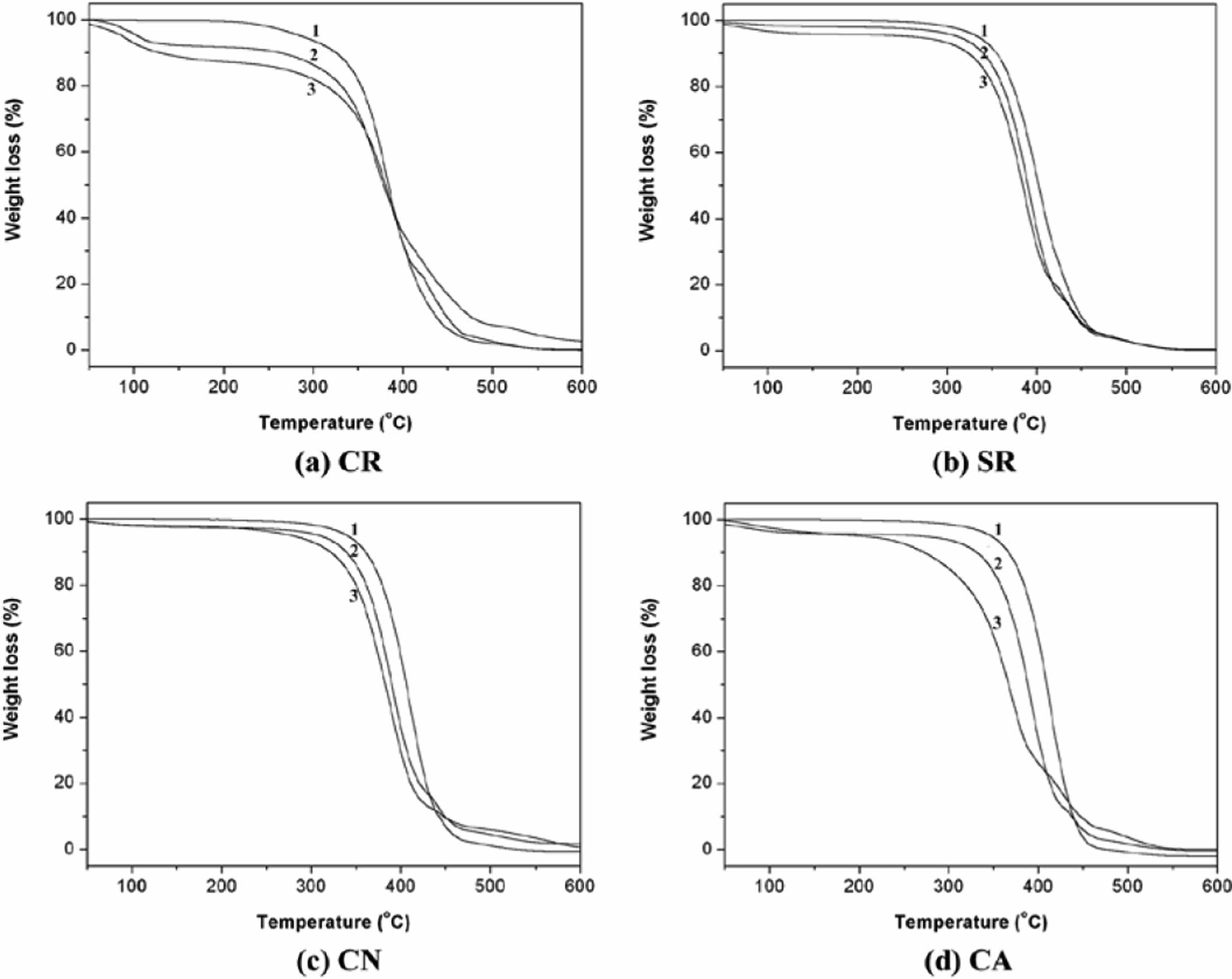
TGA of epoxidized- and methoxylated oils was carried out under similar conditions (Figure 4). The thermal degradation processes of epoxidized CR (ECR), SR (ESR), CN (ECN), and CA (ECA) are very similar, and there are three distinct and well-separated turns in the TGA curves. Volatile compounds, constituting approximately 90% of the epoxidized oils composition, decompose at temperatures around 470 ℃. The remaining 10% comprised highly viscous pyrolysis products, which underwent further decomposition up to 550 ℃. All samples were completely oxidized and volatilized upon heating to 800 ℃ (Figure 4-2).
The thermal decomposition of methoxylated CA (MCA) started at a lower temperature than that of methoxylated CR (MCR), SR (MSR), and CN (MCN), as shown in Figure 4-3, Thermally, it is stable up to 229.3 ℃ and undergoes three stages decomposition at 228.3 to 550.0 ℃ with total weight loss of 99.9%.
As depicted in Figures 4a-3, 4b-3 and 4c-3, the MCR, MSR and MCN exhibit thermal stability up to 247.4, 259.8, and 257.6 ℃, respectively. Beyond this temperature, a three-stage decomposition process (MCR: 247.4-396.6, 396.6-460.4, 460.5-550.0, MSR: 259.8-414.7, 414.7-467.8, 467.8-550.0, MCN: 257.6-418.6, 418.6-466.5, 466.5-550.0 ℃) ensues, leading to a total weight loss of approximately 96.9%-99.8% by 550 ℃. The initial weight loss of about 13.7% can be ascribed to the removal of moisture and other volatile substances present in the MCR.
TGA analysis revealed a downward shift in the onset temperature of weight loss when compared to both the pure and epoxidized oil samples. The oil's decreased thermal stability is attributed to the introduction of secondary hydroxyl groups.13 After epoxidation and methoxylation, SR, CN, and CA develop molecular structures similar to CR, with a residual double bond and secondary hydroxyl group (Scheme 1(c)). Consequently, their thermal degradation behaviors become similar to that of CR in Figure 3.
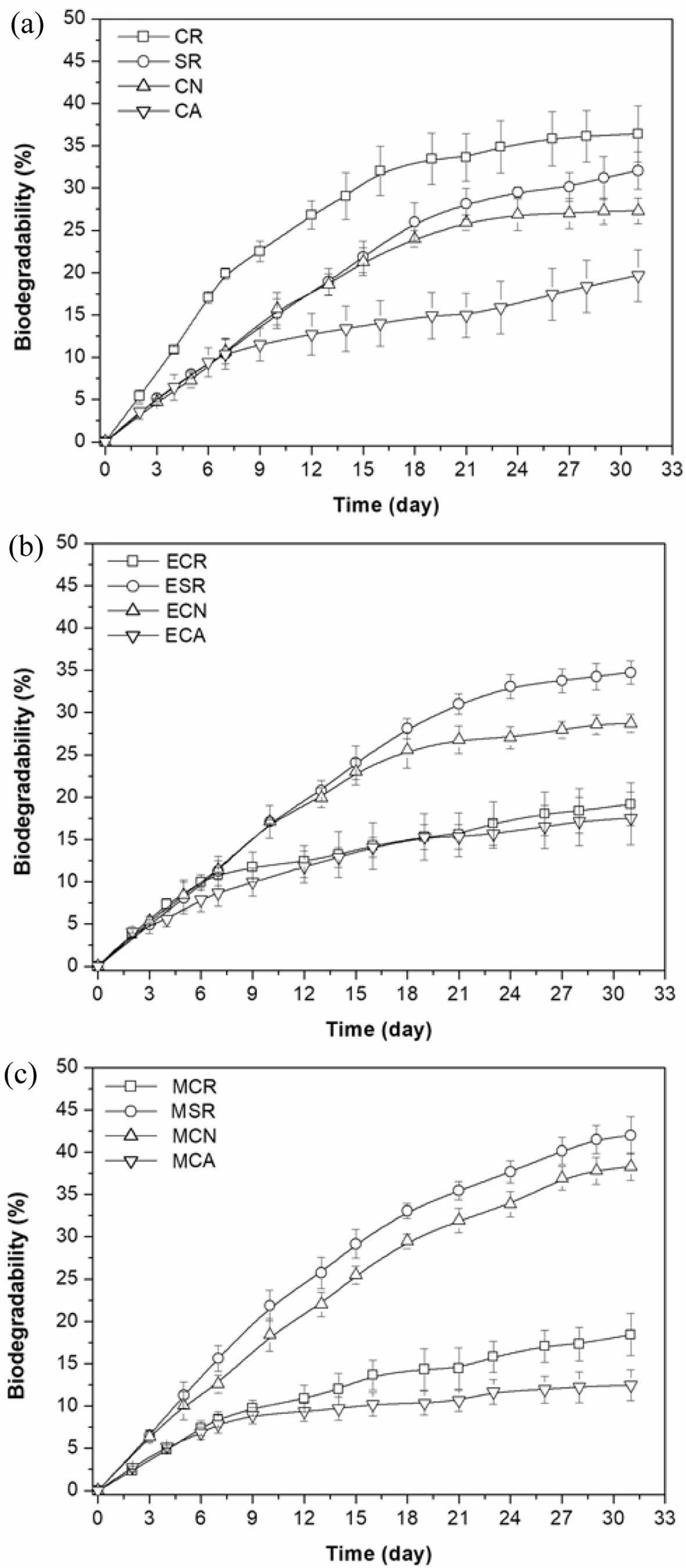
Biodegradability of Pure, Epoxidized- and Methoxylated Oils. Figure 5 illustrates the results of the Modified Sturm test, which assesses the extent of biodegradation of four vegetable oils by P. aeruginosa E7 over a specific timeframe. As depicted in Figure 5, the degradation curves follow a characteristic exponential growth profile, culminating in an asymptotic plateau. The observed initial rapid degradation rate is likely due to the concomitant increase in microbial biomass and the availability of nutrients. This observation aligns with previous studies, which have demonstrated that the addition of nutrients can significantly enhance biodegradation rates.15 However, the provision of vegetable oil as the sole carbon source is insufficient, as microorganisms necessitate a diverse range of nutrients, including phosphorus, nitrogen, sodium, iron, sulfur, and magnesium.16
The biodegradation rate of CR was the highest among the tested vegetable oils, while the CA showed the lowest biodegradation rate. SR and CN were mineralized 32.1 and 27.3%, respectively, after 31 days in Modified Sturm stock inoculated with the P. aeruginosa E7. Mineralization of CR and CA totaled 36.4 and 19.7% after 31 days of incubation, respectively. The biodegradation order is CR > SR > CN > CA.
The elevated biodegradability of CR may be attributed to the presence of hydroxyl groups, which can promote oxidative degradation by facilitating the generation of free radicals.17 The initial stage of vegetable oil biodegradation involves the enzymatic cleavage of ester linkages between glycerol and fatty acids. A diverse range of microorganisms produce enzymes, such as esterases and lipases, that catalyze this biodegradation reaction.18 Lipases catalyze the hydrolysis of ester bonds in tri-, di-, and monoacylglycerols.19 Once triacylglycerols are hydrolyzed, the released free fatty acids are transported into cells and metabolized to CO2 through β-oxidation and the incorporation of acetate into the tricarboxylic acid cycle.20
On the other hand, the highest oleic acid content CA showed the lowest biodegradability. This is because oleic acid was more saturated than linoleic acid thereby less prone to degradation. The major abundant fatty acid found in CA is oleic acid, while SR and CN are predominantly linoleic acid. CN has less linoleic acid values and slightly higher oleic acid values compared to SR (Table 1). The oxidation stability of vegetable oils is inversely proportional to the degree of fatty acid unsaturation.
Oleic acid, an 18-carbon fatty acid with a single double bond located at the ω-9 position, exhibits more oxidation stability compared to linoleic acid, an 18-carbon fatty acid with two double bonds at the ω-6 position. As an illustration, oleic acid is approximately 10 times more oxidation-resistant than linoleic acid, which, in turn, is twice as stable as linolenic acid.21
MSR and MCN were biodegraded faster and more efficiently than their corresponding parent oils (Figure 5(a), (c)). Moreover, the epoxidized SR (ESR) and epoxidized CN (ECN) have similar biodegradation behavior to the parent oils. These results indicated that P. aeruginosa E7 prefers hydroxyl fatty acids containing both a hydroxyl group and a double bond. The molecular structure of a partially methoxylated linoleic acid resembles a ricinoleic acid.
In sharp contrast, ECR, MCR, and MCA exhibited significantly different biodegradation behavior under the same conditions. The significant drop in biodegradability of ECR, MCR, and MCA could be explained by the reduction in residual unsaturation content and the increase in viscosity. CR, CA, and epoxidized CA (ECA) appear as a very pale yellow liquid at room temperature. However, ECR, MCR, and MCA are solid at room temperature and melt in the temperature range ~40 ℃. Higher viscosity can lead to decreased bioavailability and slower biodegradation.22
Preparation and Properties of PU. To obtain PU, 11.12 parts (10 wt%) by weight of PTMG and 0.32 parts by weight of TEH were added to 100 parts by weight of the PU prepolymer, mixed thoroughly, poured into a glass Petri dish, and cured at 60 ℃ for 2 h. For chain-extended PU, 11.12 parts by weight of PTMG was replaced with 11.12 (10 wt%), 17.65 (15 wt%), or 25.00 (20 wt%) parts by weight of methoxylated vegetable oil and prepared under the same conditions.
An optimal composition ratio was selected for PU to ensure both mechanical properties and biodegradability, based on our previous work.7 Compositions with over 25 wt% methoxylated oil showed a significant decrease in mechanical properties, while those below 10 wt% had little impact on biodegradability. Consequently, these compositions were excluded from this study.
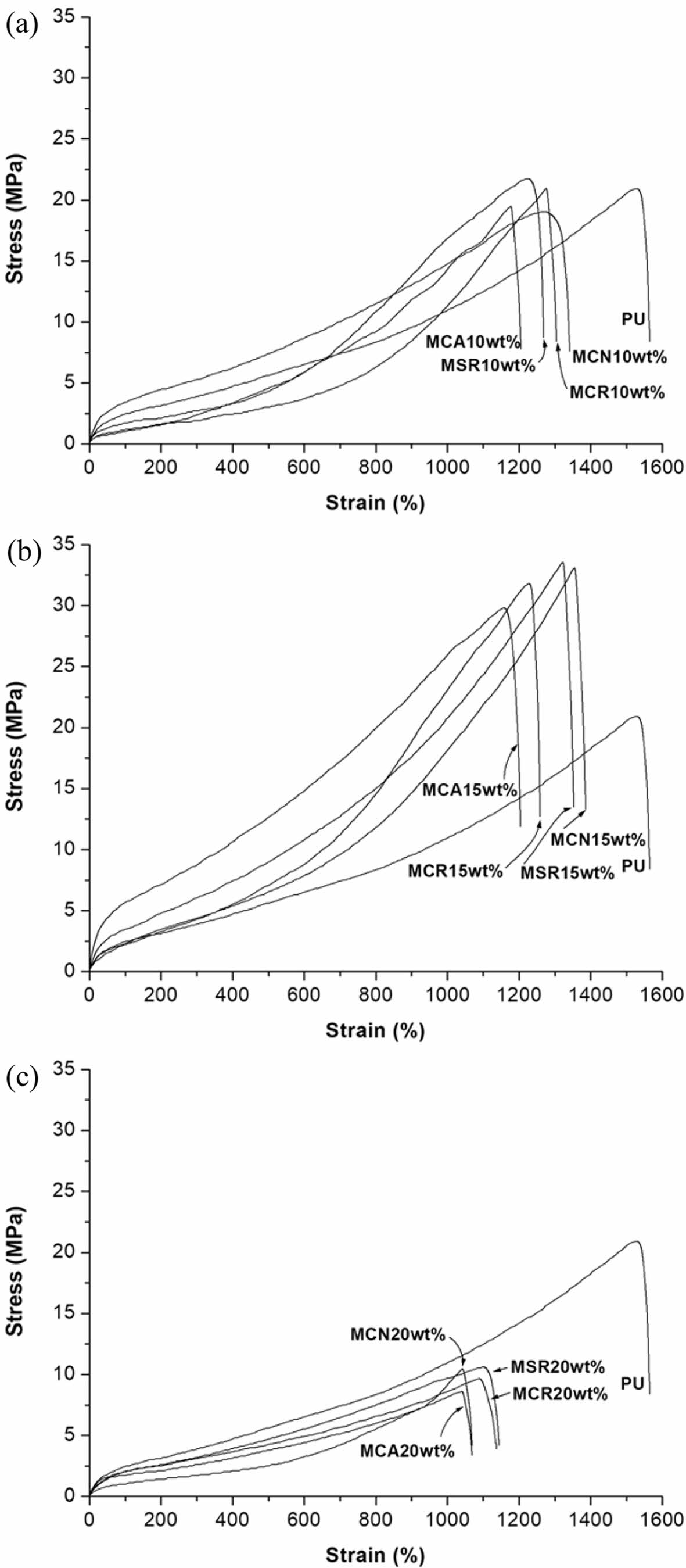
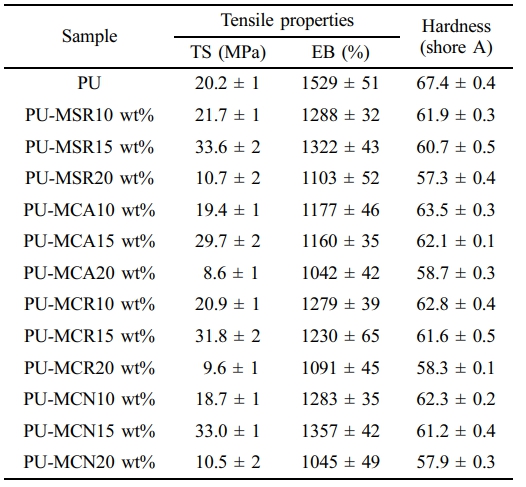
The influence of MSR, MCN, MCR, and MCA content on the mechanical properties of PU was assessed using UTM and Shore A durometer measurements. These properties are summarized in Table 2. In the case of PU chain-extended with 10 wt% methoxylated oil, the tensile strength (TS) did not show much difference from PU, while the elongation at break (EB) decreased by more than 16%. Furthermore, the TS of the prepared PU significantly increased up to 15 wt% of methoxylated vegetable oils, while it significantly decreased as the methoxylated vegetable oil content increased beyond that.
Notably, the PU-MCA system exhibited lower tensile properties compared to the other systems. When the TS of PU-MCA 15 wt% was compared with PU-MSR 15 wt%, PU-MCN 15 wt%, and MCR 15 wt%, it was found to be approximately 13, 11, and 17% lower, respectively. Likewise, EB decreased by approximately 14, 17, and 10% in the same order. The EB of PU-MCA and PU-MCR gradually decreased as the MCA and MCR contents increased to 20 wt%, respectively. On the other hand, the EB of PU-MSR and PU-MCN showed the same behavior as their TS.
The -NCO/-OH ratio decreases in the order of PU-MCA, PU-MSR, PU-MCN, and PU-MCR, with values of 2.17, 1.93, 1.92, and 1.87, respectively. This is because the hydroxyl functional group of MCA is 2.2, which is low compared to other methoxy oils, and the optimal -NCO/-OH ratio has not yet been reached.
However, the incorporation of 20 wt% methoxylated oil resulted in a rapid decrease in TS and EB for all chain-extended PUs by approximately 68% and 10%, respectively, than that of adding 15 wt%.
The hardness of PU decreased progressively with increasing content of methoxylated oil, irrespective of the specific oil type. The addition of 20 wt% of MSR, MCN, MCR, or MCA led to a substantial 7% reduction in hardness compared to the addition of 10 wt%.
In this study, five polyols (MSR, MCN, MCR, MCA, and PTMG) were used to synthesize PU. In theory, the optimal NCO/OH ratio for achieving maximum molecular weight and desired properties was found to be 1:1. PTMG, a semi-crystalline polymer, possesses two functional groups per molecule.23 This characteristic ensures the formation of linear PU during the prepolymer reaction, irrespective of the -NCO/-OH ratio (Scheme 3).
Although MSR, MCN, MCR, and MCA exhibit an amorphous structure, they contain multiple functional groups.24 When using a chain-extender with a functional group larger than 2, a partial cross-linking reaction is likely to occur even if the -NCO/-OH ratio is low. The introduction of partial cross-linking enhances rubber elasticity, resulting in an increase in TS and a decrease in hardness.
Conversely, as the –NCO/–OH ratio approaches equivalence, cross-linking density increases, and the crystalline phase decreases with higher chain-extender content. Despite a higher NCO/OH ratio in PU-MSR20 wt% (1.45), its TS decreased by 68.2% compared to PU-MSR15 wt%. The addition of 20 wt% chain-extender hindered PTMG crystallization. This loss of crystallinity resulted in a decrease in rubber elasticity, leading to softer, tackier materials.
Figure 6 illustrates representative stress-strain curves for PU as a function of methoxylated vegetable oil content. PU, PU-MSR, PU-MCN, PU-MCR, and PU-MCA exhibited predominantly elastic behavior. These materials demonstrated exceptional elongation, exceeding 1000%, and significant strain recovery upon unloading. Despite the significant deformation, the most prominent feature of the stress-strain curves is their nonlinear nature. A yield point is evident, indicating a localized, heterogeneous transition from elastic to plastic deformation.
Further, PU-MSR15, PU-MCN15, PU-MCR15, and PU-MCA15 exhibit crystallization at high strains (Figure 6(b)). It is important to note that strain-induced crystallization is particularly pronounced in certain elastomers, and filler reinforcement can mitigate or eliminate this effect entirely. As shown in Figure 6(c), when the methoxylated oil content increased to 20 wt%, the overall stress–strain curve appearance was the same as that of PU, but it can be seen that their ultimate stress and elongation were greatly reduced.
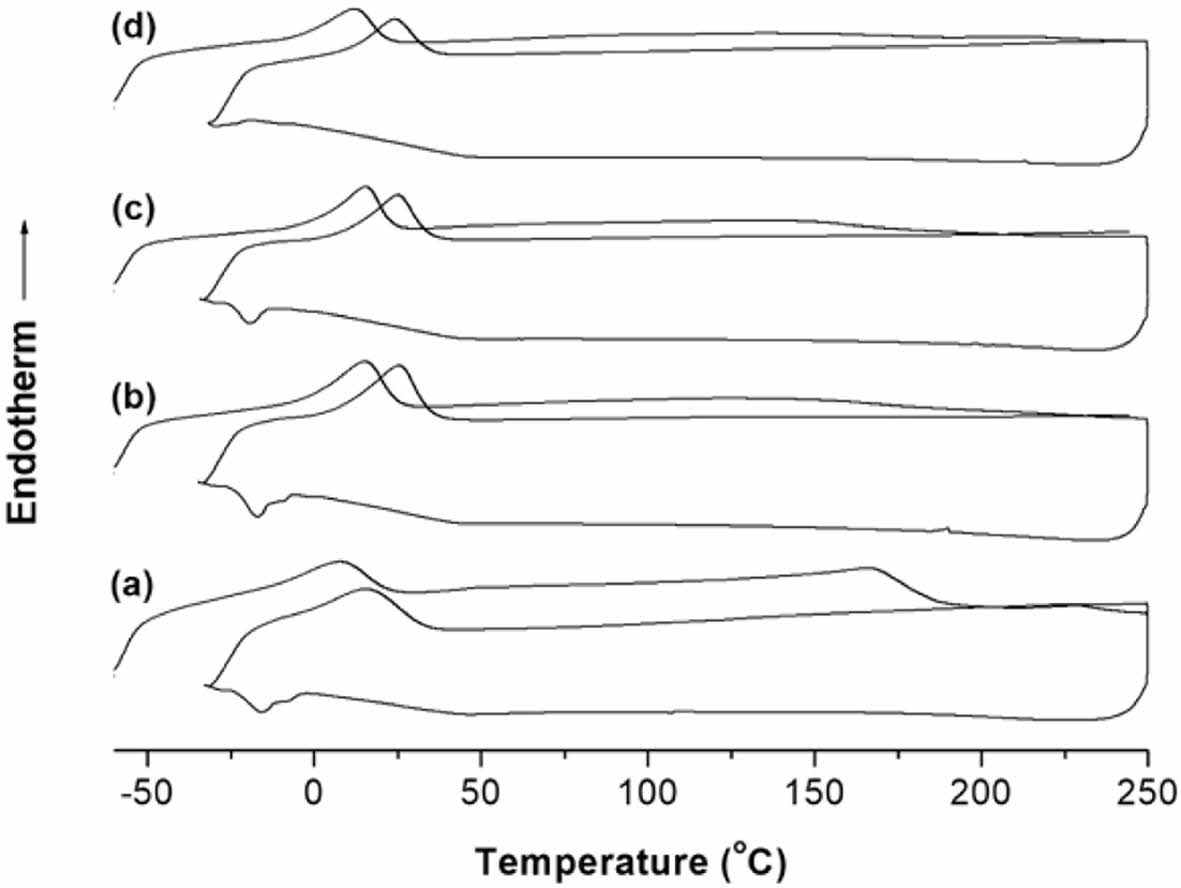
Figure 7 illustrates the differential scanning calorimetry (DSC) melting and cooling curves of PU and chain-extended PU. The first-scan DSC curve of PU revealed two distinct melting peaks at 8.0 and 167.3 ℃. These peaks were attributed to the melting of soft-segment (Tm11) and hard-segment crystals (Tm12), respectively, which formed during the curing process at 60 ℃. The Tm12 peak disappeared in the second-scan DSC thermogram. This can be attributed to the higher polarity of hard-segments compared to soft-segments, leading to rapid interaction and less stable crystal formation. Consequently, only the soft-segments, capable of forming more stable crystals, remained detectable in the second-scan curve.
From Figure 7(d), adding 20 wt% of the MSR as a chain extender causes the Tm12 peak in the first-scan DSC curve to disappear. This is because the isocyanate group at the end of the PU prepolymer reacts with the hydroxyl group in the methoxylated oil to form a cross-linking point and inhibit the interaction of the hard-segments. Therefore, as the methoxylated oil content increases, the PU molecules gradually convert into an amorphous form due to the cross-linking reaction.6
In the DSC cooling curve, PU showed split crystallization peaks at approximately -15.1 and -8.2 ℃. It is inferred that this appears in the soft-segments of the area where the chain has been extended by PTMG and the parts that have not. Likewise, PU-MSR10 wt% showed split crystallization peaks at -16.9 and -10.8 ℃. These split crystallization peaks merge into one at -19.1 when the MSR content is 15 wt% and completely disappear when the MSR content increases to 20 wt%.
Figure 8 illustrates SEM images of the cryogenic fracture surfaces of PU, PU-MSR10 wt%, and PU-MSR20 wt% samples. PU exhibited a pronounced phase-separated morphology,7 resulting from the incompatibility between PTMG soft-segments and PDI hard-segments (Figure 8(a)). A continuous phase, predominantly composed of the amorphous region of the soft-segments, is clearly visible in the SEM image. Dispersed phase particles, formed by the hard-segments and the crystalline regions of the soft-segments, are encapsulated within this continuous phase.
As shown inFigure 8(b), PU-MSR10 wt% has a similar morphology to PU, but the particle size of the dispersed phase is greatly reduced. On the other hand, PU-MSR20 wt% has a very smooth surface with no phase separation due to low crystallinity and a less regular arrangement of soft-segments (Figure 8(c)). This represents the valley-like surface that occurs when cutting highly elastic or amorphous materials.6 These results agree well with the DSC analysis.
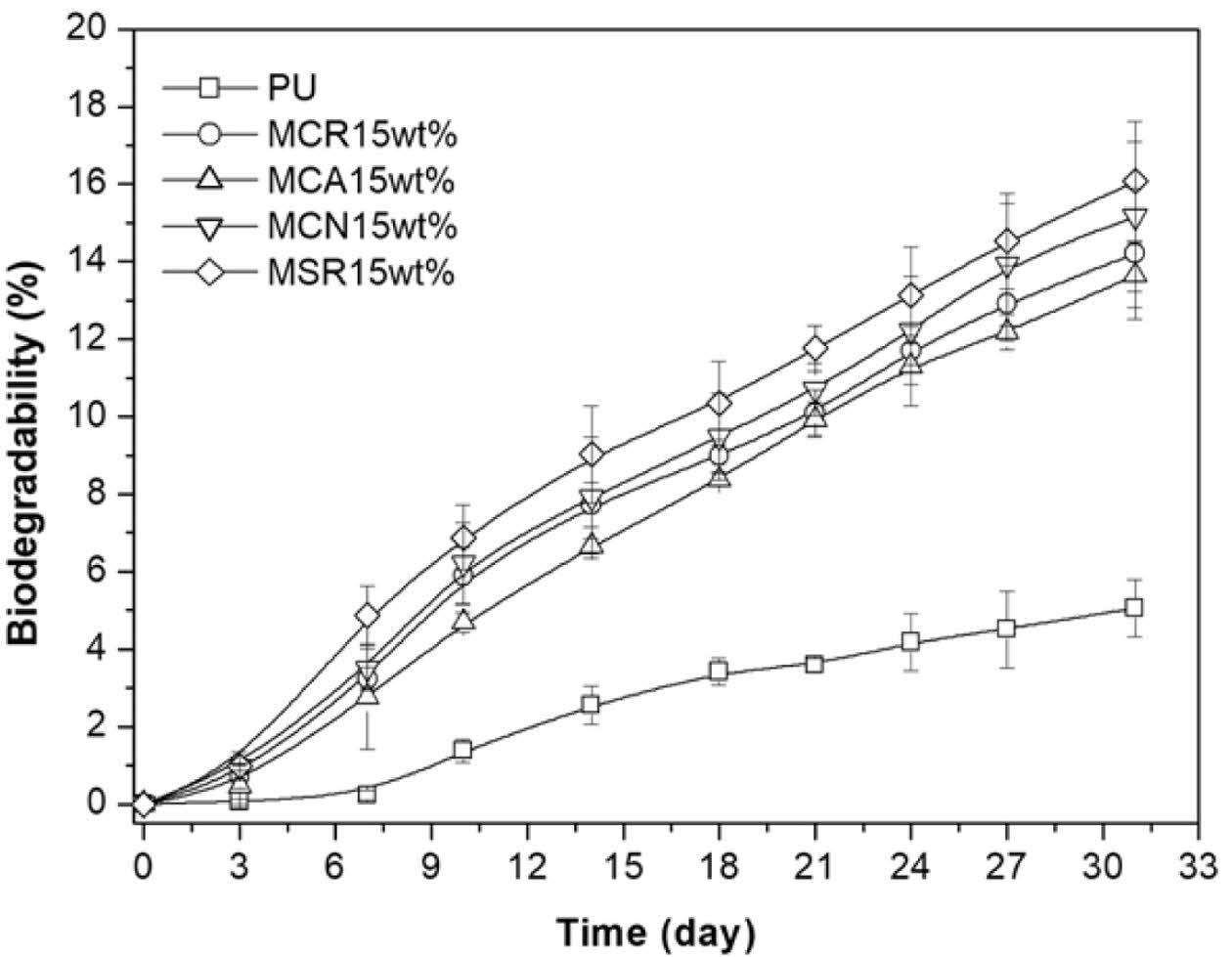
Meanwhile after 31 days of incubation, the biodegradability of PU was very low, reaching approximately 5% (Figure 9). Additionally, it can be seen that there is a time lag of 7 days during which biodegradation hardly occurs.
Hydrolysis of polymers by microbial enzymes occurs in a two-step process. An enzyme binds to the polymer substrate then subsequently catalyzes a hydrolytic cleavage. Microbial degradation of polyester type PUs proceeds preferentially by hydrolysis of ester bonds by esterase enzymes.4 The resulting low molecular weight oligomers are consumed by microorganisms and generate CO2.
The 7 days time delay that occurs in the biodegradation of PU is probably due to the time taken by the enzyme of P. aeruginosa E7 to hydrolyze the ester bond in the PTMG soft-segment.
However, the PU containing 15 wt% of MSR, MCN, MCR, and MCA showed an enhanced biodegradation rate from the initial phase of the experimental trial. Unlike PU, 15 wt% of MSR, MCN, MCR, and MCA containing PUs show no time lag for biodegradation and were asymptotically approaching biodegradation of approximately 16.1, 15.2, 14.2, and 13.6%, respectively. This is consistent with the decreasing order of TS (Table 2) and biodegradability of methoxylated oils (Figure 5(c)).
Microbial degradation of PU is influenced by factors such as crystallinity, cross-linking density, molecular orientation, and chemical group composition, which affect the accessibility of degradative enzymes.4 As shown in Figure 7, increasing MSR content leads to the merging or disappearance of PU-MSR's split crystallization peaks, indicating a decrease in PTMG crystallinity. The amorphous regions, with their loose molecular packing, are more susceptible to degradation. Conversely, crystalline regions exhibit higher resistance to degradation. Since enzymes primarily target amorphous domains, crystallinity is a crucial factor affecting biodegradability.25
|
Figure 1 1H NMR spectra of the (a) CR; (b) SR; (c) CN; (d) CA (1: pure, 2: after epoxidation, and 3: after methoxylation). |
|
Figure 2 FTIR spectra of the (a) CR; (b) SR; (c) CN; (d) CA (1: pure, 2: after epoxidation, and 3: after methoxylation). |
|
Figure 3 TGA traces of the CR, SR, CN and CA obtained by heating from 20 to 800 ℃ at a rate of 5 ℃/min under N2 atmosphere. |
|
Figure 4 TGA traces of the (a) CR; (b) SR; (c) CN; (d) CA (1: pure, 2: after epoxidation, and 3: after methoxylation). |
|
Figure 5 Modified sturm test results of the (a) pure oil (CR, SR, CN, and CA); (b) epoxidized oil (ECR, ESR, ECN, and ECA); (c) methoxylated oil (MCR, MSR, MCN, and MCA) using P. aeruginosa E7. |
|
Figure 6 Stress–strain curves of the prepared PU with methoxylated oil content of (a) 10; (b) 15; (c) 20 wt%. |
|
Figure 7 DSC thermograms of the (a) PU; (b) PU/MSR10 wt%; (c) PU/MSR15 wt%; (d) PU/MSR20 wt% samples. |

|
Figure 8 SEM image of cryogenic fracture surfaces of the (a) PU; (b) PU-MSR10 wt%; (c) PU-MSR20 wt%. |
|
Figure 9 Results of modified Sturm test of the PU (□); MCR15 wt% (○); MCA15 wt% (△); MCN15 wt% (▽); MSR15 wt% (◇) degraded by P. aeruginosa E7. |
A series of PU elastomers were synthesized using methoxylated vegetable oils (MSR, MCN, MCR, or MCA) as chain extenders. An isocyanate-terminated prepolymer, prepared from PDI and PTMG, was chain-extended with the respective methoxylated oil to yield the resulting PU. The TS of chain-extended PUs peaked when the methoxylated oil content was 15 wt% and decreased significantly thereafter. However, as the methoxylated oil content increased, their hardness gradually decreased.
After incubation for 31 days, the biodegradability of PU was very low, reaching approximately 5%. Additionally, it can be seen that there is a lag of 7 days during which biodegradation hardly occurs. However, the chain-extended PU with 15 wt% of MSR, MCN, MCR, and MCA showed improved biodegradation rates than pure PU, with no time delay for biodegradation. This is consistent with the decreasing order of TS of chain-extended PU and biodegradability of methoxylated oil.
SEM analysis revealed that PU-MSR10 wt% exhibited significant phase separation between hard- and soft-segments, similar to pure PU. Conversely, PU-MSR20 wt% displayed a smooth surface without discernible phase boundaries. DSC analysis indicated that increasing MSR content led to the merging or disappearance of the split crystallization peaks of PU-MSR, suggesting a reduction in the PTMG crystalline phase. These findings support the notion that elevated MSR content diminishes the PTMG crystalline phase and compromises rubber elasticity.
- 1. Zhang, J.; Tang, J. J.; Zhang, J. X. Polyols Prepared from Ring-Opening Epoxidized Soybean Oil by a Castor Oil-Based Fatty Diol. Int. J. Polym. Sci. 2015, 2015, 529235.
-
- 2. Rosenboom, J.-G.; Langer, R.; Traverso, G. Bioplastics for a Circular Economy. Nat. Rev. Mater. 2022, 7, 117-137.
-
- 3. Salimon, J.; Abdullah, B. M.; Yusop, R. M.; Salih, N. Synthesis, Reactivity and Application Studies for Different Biolubricants. Chem. Cent. J. 2014, 8, 16.
-
- 4. Su, T.; Zhang, T.; Liu, P.; Bian, J.; Zheng, Y.; Yuan, Y.; Li, Q.; Liang, Q.; Qi, Q. Biodegradation of Polyurethane by the Microbial Consortia Enriched From Landfill. Int. J. Polym. Sci. 2023, 107, 1983-1995.
-
- 5. Pongmuksuwan, P.; Harnnarongchai, W. Synthesis and Characterization of Soft Polyurethane for Pressure Ulcer Prevention. Polym. Test. 2022, 112, 107634.
-
- 6. Datta, J.; Kosiorek, P.; Włoch, M. Synthesis, Structure and Properties of Poly(ether-urethane)s Synthesized Using a Tri-functional Oxypropylated Glycerol as a Polyol. J. Therm. Anal. Calorim. 2017, 128, 155-167.
-
- 7. Jeon, H. J.; Kim, M. N.; Park, E. S. Thermal, Mechanical and Biodegradation Properties of Pure, Epoxidized and Methoxylated Castor oil Based Polyurethane. Plast. Rubber. Compos. 2016, 45, 1-8.
-
- 8. Jeon, H. J.; Kim, M. N. Functional Analysis of Alkane Hydroxylase Systems Derived from Pseudomonas aeruginosa E7 for Low Molecular Weight Polyethylene Biodegradation. Int. Biodeter. Biodegr. 2015, 103, 141-146.
-
- 9. Meshram, P. D.; Puri, R. G.; Patil, H. V. Epoxidation of Wild Safflower (Carthamus oxyacantha) Oil with Peroxy Acid in Presence of Strongly Acidic Cation Exchange Resin IR-122 as Catalyst. Int. J. ChemTech Res. 2011, 3, 1152-1163.
- 10. Parés, X. P. X.; Bonnet, C.; Morin, O. Synthesis of New Derivatives from Vegetable Oil Methyl Esters via Epoxidation and Oxirane Opening. In Recent Developments in the Synthesis of Fatty Acid Derivatives; Knothe, G.; Derksen, J. T. P., Eds.; AOCS Publications: Champaign, 1999; pp. 141-156.
-
- 11. Souza, A. G.; Santos, J. C. O; Conceição, M. M.; Silva, M. C. D.; Prasad, S. A Thermoanalytic and Kinetic Study of Sunflower Oil. Braz. J. Chem. Eng. 2004, 21, 265-273.
-
- 12. Silva, J. A. C. Biodegradable Lubricants and Their Production Via Chemical Catalysis. In Tribology - Lubricants and Lubrication; Kuo, C.-H., Eds.; Intech: Rijeka, 2011; pp.185-187.
-
- 13. Murru, C.; Badía-Laíño, R.; Díaz-García, M. E. Oxidative Stability of Vegetal Oil-Based Lubricants. ACS Sustainable Chem. Eng. 2021, 9, 1459-1476.
-
- 14. Sruthi, R.; Kumar, K. R.; Sirisha, G. Determines of Physico-chemical Properties of Castor Biodiesel: A Potentials Alternate to Conventional Diesel. IJARET 2013, 4, 101-107.
- 15. Bharath, P.; Elavaras, N.; Mohana, S.; Sundaram, N. Studies on Rate of Biodegradation of Vegetable (coconut) Oil by Using Pseudomonas aeruginosa. Int. J. Environ. Biol. 2012, 2, 12-19.
- 16. Alsulami, A. A.; Altaee, A. M. R.; FAl-Kanany, F. N. A. Improving oil Biodegradability of Aliphatic Crude oil Fraction by Bacteria From Oil Polluted Water. Afr. J. Biotechnol. 2014, 13, 1243-1249.
-
- 17. Salih, N.; Salimon, J.; Yousif, E.; Abdullah, B. M. Biolubricant Basestocks from Chemically Modified Plant Oils: Ricinoleic Acid Based-tetraesters. Chem. Cent. J. 2013, 7, 128.
-
- 18. Broekhuizen, P.; Theodori, D.; Blanch, K.; Ullmer, S. Lubrication in Inland and Coastal Water Activities; AA Balkema Publishers: Tokyo, 2003.
- 19. Saifudin, N.; Chua, K. H. Biodegradation of Lipid-rich Waste Water by Combination of Microwave Irradiation and Lipase Immobilized on Chitosan. Biotech. 2006, 5, 315-323.
-
- 20. Ratledge, C. Biochemistry of Microbial Degradation; Kluwer Academic Publishers: Dordrecht, 1994.
- 21. Li, J.; Liu, J.; Sun, X.; Liu, Y. The Mathematical Prediction Model for the Oxidative Stability of Vegetable Oils by the Main Fatty Acids Composition and Thermogravimetric Analysis. LWT 2018, 96, 51-57.
-
- 22. Haus, F.; Boissel, O.; Junter, G.-A. Primary and Ultimate Biodegradabilities of Mineral Base Oils and Their Relationships with Oil Viscosity. Inter. Biodet. Biodeg. 2004, 54, 189-192.
-
- 23. Raftopoulos, K. N.; Janowski, B.; Apekis, L.; Pielichowski, K.; Pissis, P. Molecular Mobility and Crystallinity in Polytetramethylene Ether Glycol in the Bulk and as Soft Component in Polyurethanes. Eur. Polym. J. 2011, 47, 2120-2133.
-
- 24. Petrović, Z. S.; Guo, A.; Zhang, W. Structure and Properties of Polyurethanes Based on Halogenated and Nonhalogenated Soy–polyols. J. Polym. Sci. A: Polym. Chem. 2000, 38, 4062-4069.
-
- 25. Limsukon, W.; Rubino, M.; Rabnawaz, M.; Lim, L.-T.; Auras, R. Hydrolytic Degradation of Poly(lactic acid): Unraveling Correlations Between Temperature and the Three Phase Structures. Polym. Deg. Stab. 2023, 217, 110537.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2025 Impact Factor : 1.0
- Indexed in SCIE
 This Article
This Article
-
2025; 49(3): 285-295
Published online May 25, 2025
- 10.7317/pk.2025.49.3.285
- Received on Oct 12, 2024
- Revised on Dec 20, 2024
- Accepted on Jan 7, 2025
Services
- Full Text PDF
- Abstract
- ToC
- Conflict of Interest
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Young Jun Kim , and Eun-Soo Park*
-
Department of Food & Biotechnology, Korea University Sejong Campus, 2511, Sejong-ro, Jochiwon-eup, Sejong-si 30019, Korea
*Intechnology Co., Ltd., 861-46, Poseungjangan-ro, Jang-an-ri, Jangan-myeon, Hwaseong-si, Gyeonggi-do 18586, Korea - E-mail: yk46@korea.ac.kr, t2phage@hitel.net
- ORCID:
0000-0003-1447-7857, 0000-0002-4150-7401
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr